Model based visualization of ancient DNA damage using aRchaic
Kushal K Dey
Department of Statistics, University of ChicagoHussein Al-Asadi
Department of Ecology and Evolution, University of ChicagoSource:
vignettes/archaic.Rmd
archaic.Rmd.
Introduction
aRchaic is an R package that performs model based clustering and visualization of ancient and modern DNA samples (see archaic_plot() section for example visualization). This model can be used to identify distinct patterns of DNA damage and is capable of reflecting relative rates of contamination in contaminated aDNA samples.
Installation
First and foremost, the user is required to install
aRchaic requires R version to be (>= 3.4). If your R version is lower, please upgrade.
And Python packages,
Upon completion of these steps, start a new R session and install aRchaic:
install.packages("remotes") remotes::install_github("kkdey/aRchaic")
Now you should be able to load aRchaic into R.
library(aRchaic)
To ensure reproducible results, we set the seed:
set.seed(1)
Generate aRchaic input (MFF file)
For every mismatch in a BAM file with respect to a reference genome, aRchaic records various features of the mismatch - mismatch type, flanking bases, strand break base, strand and position of mismatch from the ends of read - into a .csv file which we call the Mismatch Feature Format (MFF) file.
A typical example MFF file looks like the following
mff <- read.csv(system.file("extdata","ancients","NE2_subsampled.csv",package = "aRchaic"), header = FALSE) head(mff)
## V1 V2 V3 V4 V5 V6 V7
## 1 AC->TC 17 48 A A - 1557694
## 2 AG->AC 47 9 A T + 4607735
## 3 TG->AT 93 1 A C - 5166724
## 4 TC->TT 16 50 G G - 4581952
## 5 AC->TG 48 31 A C + 207160
## 6 GG->AG 33 5 G G - 2912042The first column represents mismatch type and flanking bases : (left flank base)(mismatch)(right flank base) <-> (A)(C->T)(C). The second and third columns represent the position of mismatch from 5’ and 3’ ends of reads after mapped to reference. The 4th (5th) columns represent the base 1 base upstream (downstream) from 5’ (3’) end of read respectively. The 6th column contains information of which strand the read comes from, and the 7th column is an identifier for the read containing the mismatch.
We provide a script to generate the MFF files from the BAM/SAM files here. This linked folder also contains a small example BAM file for testing. The simple two-line code to create the MFF file from this BAM file is as follows
samtools index example.bam
python generate_summary_bams.py -b example.bam -f /path/to/reference/FASTA/hs37d5
.fa -o example_mff.csvarchaic_prepare()
The user is expected to organize the MFF files in seperate folders per study. Here, we provide an example with two studies, one with 5 ancient samples derived from Gamba et. al. 2014 and a second study with 5 modern samples derived from the 1000 Genomes Project. These two folders are automatically downloaded and saved under inst/extdata/moderns and inst/extdata/ancients when aRchaic is installed.
- Modern study :
moderns_dir <- system.file("extdata","moderns", package = "aRchaic") list.files(moderns_dir, pattern = ".csv")
## [1] "NA19788_subsampled.csv" "NA19789_subsampled.csv" "NA19792_subsampled.csv"
## [4] "NA19794_subsampled.csv" "NA19795_subsampled.csv"- Ancient study :
ancients_dir <- system.file("extdata","ancients", package = "aRchaic") list.files(ancients_dir, pattern = ".csv")
## [1] "NE2_subsampled.csv" "NE3_subsampled.csv" "NE4_subsampled.csv"
## [4] "NE5_subsampled.csv" "NE6_subsampled.csv"Next we prepare the input data to aRchaic model from the above data folders.
out <- archaic_prepare(dirs = c(moderns_dir, ancients_dir))
## Reading file /private/var/folders/9b/ck4lp8s140lcksryyh4dppdr0000gn/T/RtmpzwN1lw/temp_libpath708d3f32698b/aRchaic/extdata/moderns/NA19788_subsampled.csv
## Reading file /private/var/folders/9b/ck4lp8s140lcksryyh4dppdr0000gn/T/RtmpzwN1lw/temp_libpath708d3f32698b/aRchaic/extdata/moderns/NA19789_subsampled.csv
## Reading file /private/var/folders/9b/ck4lp8s140lcksryyh4dppdr0000gn/T/RtmpzwN1lw/temp_libpath708d3f32698b/aRchaic/extdata/moderns/NA19792_subsampled.csv
## Reading file /private/var/folders/9b/ck4lp8s140lcksryyh4dppdr0000gn/T/RtmpzwN1lw/temp_libpath708d3f32698b/aRchaic/extdata/moderns/NA19794_subsampled.csv
## Reading file /private/var/folders/9b/ck4lp8s140lcksryyh4dppdr0000gn/T/RtmpzwN1lw/temp_libpath708d3f32698b/aRchaic/extdata/moderns/NA19795_subsampled.csv
## Reading file /private/var/folders/9b/ck4lp8s140lcksryyh4dppdr0000gn/T/RtmpzwN1lw/temp_libpath708d3f32698b/aRchaic/extdata/ancients/NE2_subsampled.csv
## Reading file /private/var/folders/9b/ck4lp8s140lcksryyh4dppdr0000gn/T/RtmpzwN1lw/temp_libpath708d3f32698b/aRchaic/extdata/ancients/NE3_subsampled.csv
## Reading file /private/var/folders/9b/ck4lp8s140lcksryyh4dppdr0000gn/T/RtmpzwN1lw/temp_libpath708d3f32698b/aRchaic/extdata/ancients/NE4_subsampled.csv
## Reading file /private/var/folders/9b/ck4lp8s140lcksryyh4dppdr0000gn/T/RtmpzwN1lw/temp_libpath708d3f32698b/aRchaic/extdata/ancients/NE5_subsampled.csv
## Reading file /private/var/folders/9b/ck4lp8s140lcksryyh4dppdr0000gn/T/RtmpzwN1lw/temp_libpath708d3f32698b/aRchaic/extdata/ancients/NE6_subsampled.csvThe output out should be a list with 2 elements - corresponding to 2 study folders. Each element is a matrix with samples along the rows, coded mismatch features along columns, and a cell in the matrix record counts of the number of times a coded mismatch pattern occurs in a sample.
We provide here the modern MFF files used in Figure 2 of our paper for reproducing the analysis.
archaic_fit()
aRchaic fits a mixed membership model on the prepared mismatch feature data from archaic_prepare() that allows each sample to have partial memberships in more than one cluster where the clusters represent distinct mismatch feature profiles.
We first choose a directory - output_dir - to save the model output and visualization.
Next we run the archaic_fit function to fit the mixed membership model. We choose the number of clusters \(K=2\)
output_dir <- tempdir() model <- archaic_fit(out, K = 2, output_dir = output_dir)
## The data is read as a list of matrices - processed by archaic_prepare()## Fitting the Grade of Membership Model - independent version - due to Y. Shiraichi and M. Stephens##
## Estimating on a 10 samples collection.
## Fitting the 2 clusters/topics model.
## log posterior increase: 21212.8, done.the output model consists of
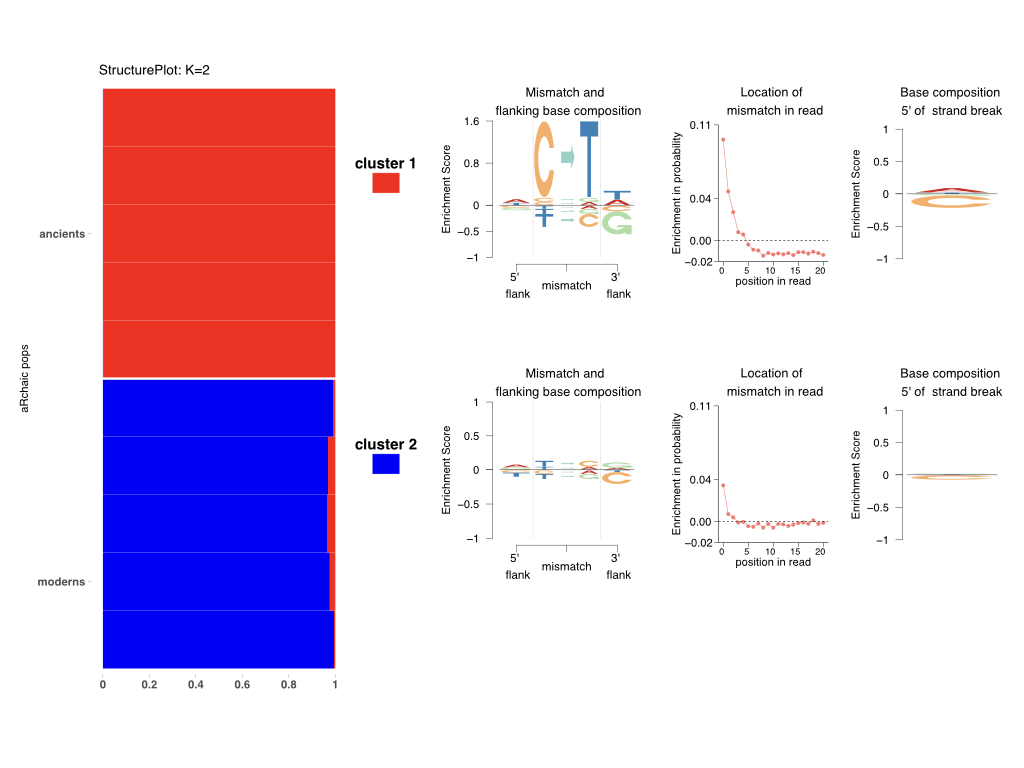
- omega : the grades of membership to be plotted by stacked bar chart
- theta : cluster profiles - with each column a probability distribution on coded mismatch features
- assessment : assessment of model fit : BIC and loglikelihood
archaic_plot()
The model output, in particular omega and theta can be visualized using the archaic_plot() command.
archaic_plot(model, output_dir = output_dir, background = "modern")
Once the run finishes (a few seconds), go to the output directory output_dir. You should see there is a structure.pdf file that represents the cluster memberships in omega in a stacked bar chart (see Fig 1 left).
Also, the output_dir should contain for each \(k=1,2\), a logo_clus_k.pdf file visualizing the mismatch profile for each cluster as in theta. If background = "modern" in archaic_plot code above, this reresentation will plot enrichment of the mismatch profiles with respect to a modern background.
The following is a summary of these visualizations