GTEx - variation in CorShrink matrices
Kushal K Dey
2/23/2018
We track the variation in the tissue-tissue correlation matrices for different genes.
library(corrplot)
## corrplot 0.84 loaded
library(gridExtra)
common_samples <- get(load("../shared_output/common_samples.rda"))
gene_names <- as.character(read.table(file = "../shared_output/GTEX_V6/gene_names_GTEX_V6.txt")[,1])
gene_names_1 <- as.character(sapply(gene_names, function(x) return(strsplit(x, "[.]")[[1]][1])))
person_label=read.table("../shared_output/GTEX_V6/person_identifier_labels_with_numbers.txt");
samples_id <- read.table(file = "../shared_output/GTEX_V6/samples_id.txt")[,1]
samples_person <- sapply(samples_id, function(x) return(paste0(strsplit(as.character(x), "-")[[1]][1:2], collapse ="-")))
tissue_labels <- read.table(file = "../shared_output/GTEX_V6/samples_id.txt")[,3]
unique_persons <- unique(samples_person)
unique_tissues <- unique(tissue_labels)
U <- unique(tissue_labels)
order_index <- get(load("../shared_output/order_index.rda"))
Correlation matrices
cor_data <- get(load("../shared_output/cor_tissues_non_ash_voom_pearson.rda"))
tissuewide_corshrink_nonmode <- get(load("../shared_output/tissuewide_pearson_halfuniform_tissuewide_non_mode.rda"))
genewide_corshrink_nonmode <- get(load("../shared_output/genewide_ash_out_tissue_mat_halfuniform_non_mode.rda"))
HBB
name <- "ENSG00000244734"
par(mfrow=c(1,3))
numg <- grep(name, gene_names_1)
col2 <- c("blue", "white", "red")
cor2mat <- cor_data[order_index, order_index, numg] + diag(1, dim(cor_data)[1])
rownames(cor2mat) <- U[order_index]
colnames(cor2mat) <- U[order_index]
g1 <- corrplot(cor2mat, diag = FALSE,
col = colorRampPalette(col2)(200),
tl.pos = "td", tl.cex = 0.5, tl.col = "black",
rect.col = "white",na.label.col = "white",
method = "color", type = "upper")
cor3mat <- tissuewide_corshrink_nonmode[order_index, order_index, numg]
rownames(cor3mat) <- U[order_index]
colnames(cor3mat) <- U[order_index]
g2 <- corrplot(cor3mat, diag = FALSE,
col = colorRampPalette(col2)(200),
tl.pos = "td", tl.cex = 0.5, tl.col = "black",
rect.col = "white",na.label.col = "white",
method = "color", type = "upper")
cor4mat <- genewide_corshrink_nonmode[order_index, order_index, numg]
rownames(cor4mat) <- U[order_index]
colnames(cor4mat) <- U[order_index]
g3 <- corrplot(cor4mat, diag = FALSE,
col = colorRampPalette(col2)(200),
tl.pos = "td", tl.cex = 0.5, tl.col = "black",
rect.col = "white",na.label.col = "white",
method = "color", type = "upper")

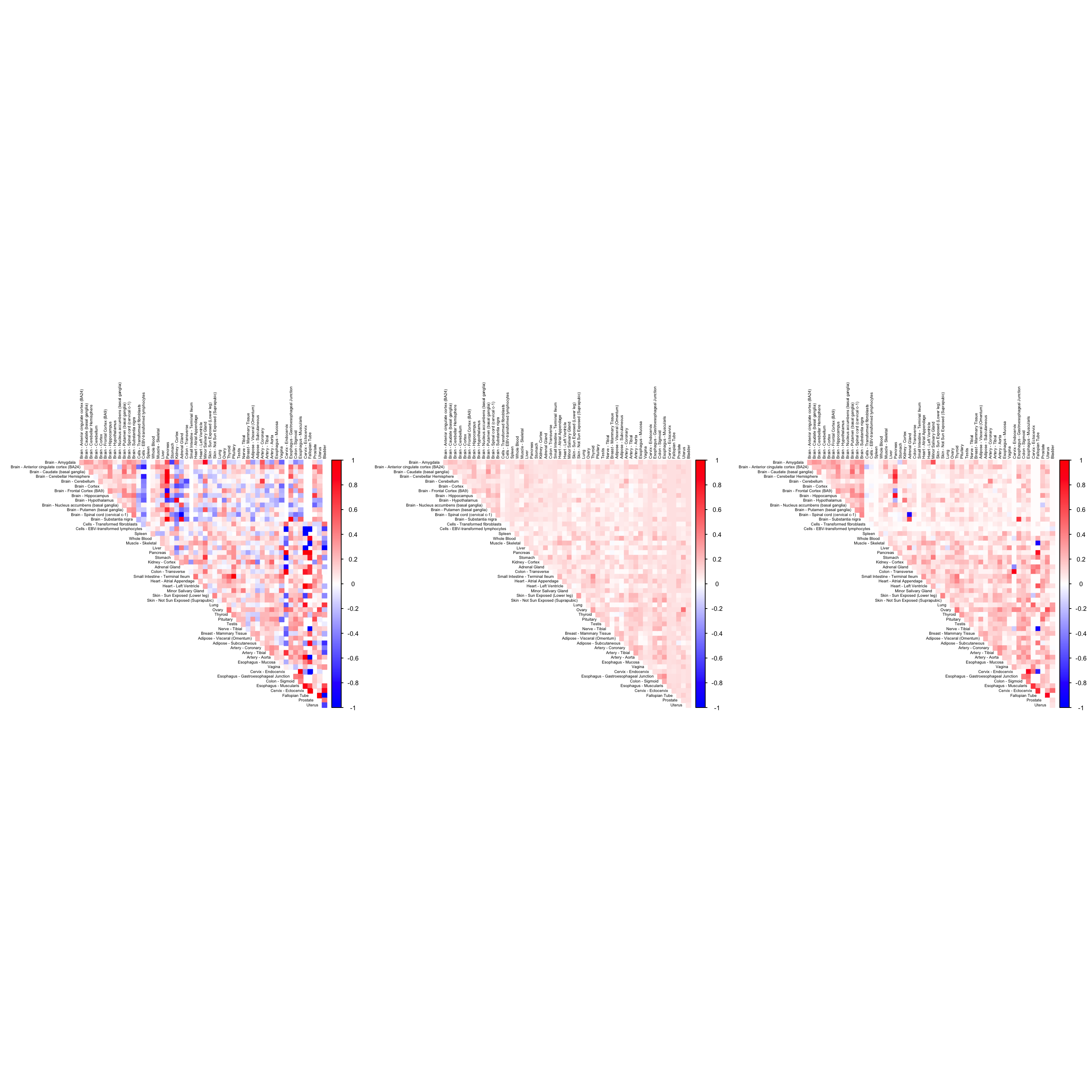
MTURN
name <- "ENSG00000180354"
numg <- grep(name, gene_names_1)
cor2mat <- cor_data[order_index, order_index, numg] + diag(1, dim(cor_data)[1])
rownames(cor2mat) <- U[order_index]
colnames(cor2mat) <- U[order_index]
par(mfrow=c(1,3))
g4 <- corrplot(cor2mat, diag = FALSE,
col = colorRampPalette(col2)(200),
tl.pos = "td", tl.cex = 0.5, tl.col = "black",
rect.col = "white",na.label.col = "white",
method = "color", type = "upper")
cor3mat <- tissuewide_corshrink_nonmode[order_index, order_index, numg]
rownames(cor3mat) <- U[order_index]
colnames(cor3mat) <- U[order_index]
g5 <- corrplot(cor3mat, diag = FALSE,
col = colorRampPalette(col2)(200),
tl.pos = "td", tl.cex = 0.5, tl.col = "black",
rect.col = "white",na.label.col = "white",
method = "color", type = "upper")
cor4mat <- genewide_corshrink_nonmode[order_index, order_index, numg]
rownames(cor4mat) <- U[order_index]
colnames(cor4mat) <- U[order_index]
g6 <- corrplot(cor4mat, diag = FALSE,
col = colorRampPalette(col2)(200),
tl.pos = "td", tl.cex = 0.5, tl.col = "black",
rect.col = "white",na.label.col = "white",
method = "color", type = "upper")
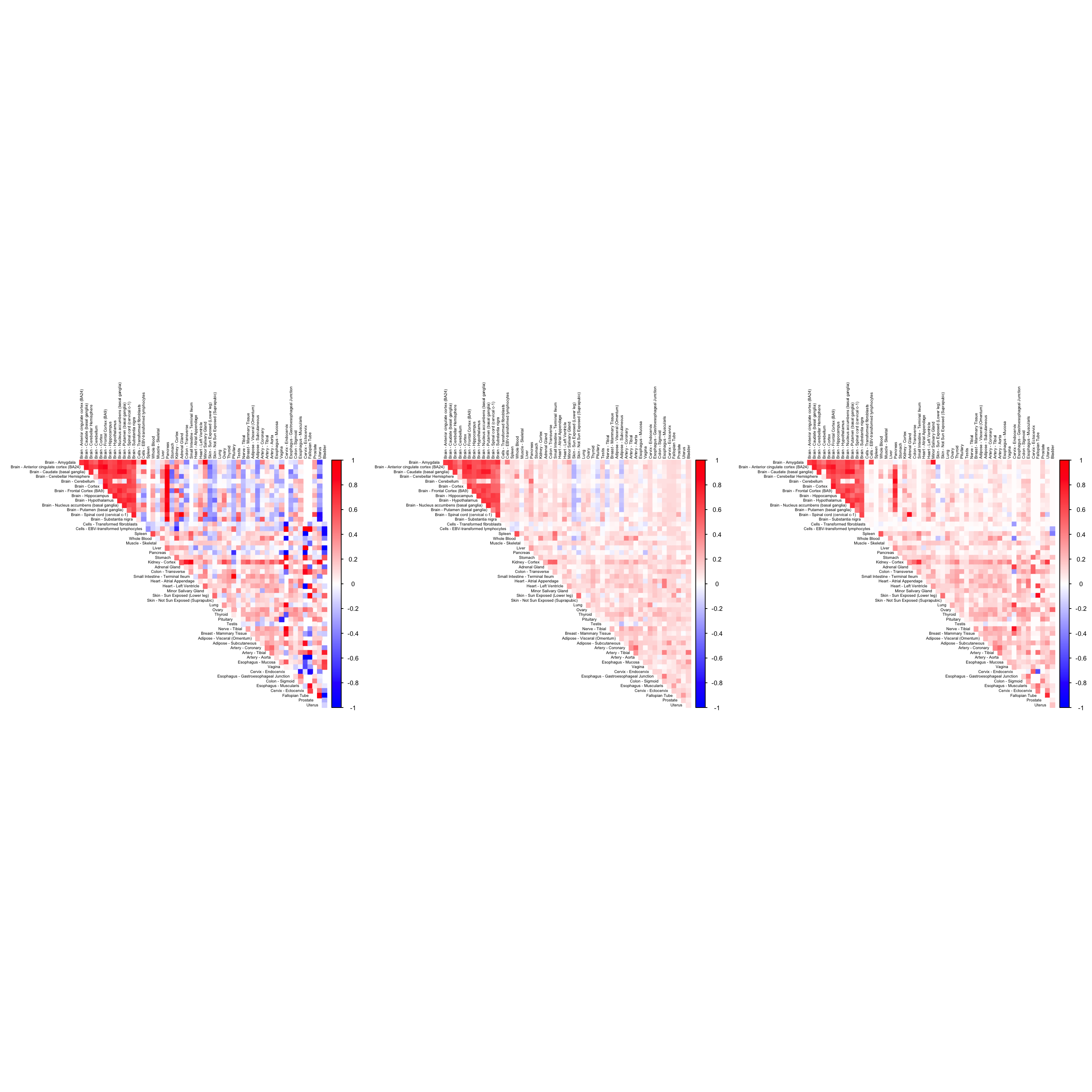
VSIR
name <- "ENSG00000107738"
par(mfrow=c(1,3))
numg <- grep(name, gene_names_1)
cor2mat <- cor_data[order_index, order_index, numg] + diag(1, dim(cor_data)[1])
rownames(cor2mat) <- U[order_index]
colnames(cor2mat) <- U[order_index]
g10 <- corrplot(cor2mat, diag = FALSE,
col = colorRampPalette(col2)(200),
tl.pos = "td", tl.cex = 0.5, tl.col = "black",
rect.col = "white",na.label.col = "white",
method = "color", type = "upper")
cor3mat <- tissuewide_corshrink_nonmode[order_index, order_index, numg]
rownames(cor3mat) <- U[order_index]
colnames(cor3mat) <- U[order_index]
g11 <- corrplot(cor3mat, diag = FALSE,
col = colorRampPalette(col2)(200),
tl.pos = "td", tl.cex = 0.5, tl.col = "black",
rect.col = "white",na.label.col = "white",
method = "color", type = "upper")
cor4mat <- genewide_corshrink_nonmode[order_index, order_index, numg]
rownames(cor4mat) <- U[order_index]
colnames(cor4mat) <- U[order_index]
g12 <- corrplot(cor4mat, diag = FALSE,
col = colorRampPalette(col2)(200),
tl.pos = "td", tl.cex = 0.5, tl.col = "black",
rect.col = "white",na.label.col = "white",
method = "color", type = "upper")
This R Markdown site was created with workflowr