GTEx Robocov demo and RoboSpan
Kushal K. Dey and Alkes Price
11/23/2019
Last updated: 2020-03-08
workflowr checks: (Click a bullet for more information)-
✖ R Markdown file: uncommitted changes
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can runwflow_publishto commit the R Markdown file and build the HTML. -
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(20190721)The command
set.seed(20190721)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 47185fd
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .DS_Store Ignored: .Rhistory Ignored: .Rproj.user/ Ignored: docs/.DS_Store Ignored: draft/ Ignored: output/output_sparse/ Untracked files: Untracked: .gitignore~ Untracked: analysis/band_prec_robocov.Rmd Untracked: analysis/blood_corspan_robospan_probospan.Rmd Untracked: analysis/corspan_robospan_probospan.Rmd Untracked: analysis/diff_robospan_examples.Rmd Untracked: analysis/eigenvalues_hub.Rmd Untracked: analysis/figure2.Rmd Untracked: analysis/gtex_analysis_robocov_examples.Rmd Untracked: analysis/gtex_pRobocov_pRobospan.Rmd Untracked: analysis/gtex_predictive_robocov.Rmd Untracked: analysis/gtex_rank_imputation.Rmd Untracked: analysis/gtex_robocov.Rmd Untracked: analysis/gtex_robocov_ex_robospan.Rmd Untracked: analysis/housekeeping_PPI_MR_enrichment.Rmd Untracked: analysis/hub_robocov.Rmd Untracked: analysis/mean_robocov.Rmd Untracked: analysis/pLI_shet_robospan_probospan.Rmd Untracked: analysis/predictive_analytics_gtex.Rmd Untracked: analysis/robocov_demo.Rmd Untracked: analysis/robospan_blood_compare.Rmd Untracked: analysis/robospan_corr.Rmd Untracked: analysis/summary_correlation_structure.Rmd Untracked: analysis/supp_figure_simulation.Rmd Untracked: analysis/toeplitz_robocov.Rmd Untracked: code/ robocov_sim.R Untracked: code/FDR_Robospan.R Untracked: code/Joints_Nov24.R Untracked: code/Untitled.R Untracked: code/annot_size.R Untracked: code/band_prec_ocor_sim.R Untracked: code/band_prec_pcor_sim.R Untracked: code/band_prec_sim.R Untracked: code/band_prec_sim2.R Untracked: code/bandprec_cor_sim.R Untracked: code/baseline_strategies.R Untracked: code/corr_span.R Untracked: code/correlation_gene_scores.R Untracked: code/gene_score_all_strategies.R Untracked: code/gtex_robocov.R Untracked: code/gtex_robocov_precision.R Untracked: code/hub_prec_cor_sim.R Untracked: code/hub_sim.R Untracked: code/hub_sim2.R Untracked: code/hub_sim_sparse.R Untracked: code/joint_robocov_model.R Untracked: code/joints_robocov.R Untracked: code/many_tau_star.R Untracked: code/maxHiCgene.R Untracked: code/meta_enrich.R Untracked: code/meta_single_enrich.R Untracked: code/postprocess_hub_toeplitz.R Untracked: code/predictive_analytics_hub.R Untracked: code/predictive_analytics_toeplitz.R Untracked: code/robocov_gtex.R Untracked: code/robocov_gtex_blood.R Untracked: code/robocov_gtex_brain.R Untracked: code/sim_results.R Untracked: code/single_tau_star.R Untracked: code/toeplitz_prec_cor_sim.R Untracked: code/toeplitz_sim.R Untracked: code/toeplitz_sim2.R Untracked: code/toeplitz_sim_sparse.R Untracked: data/Cor_pairwise_all_genes.rda Untracked: data/Gene_Scores/ Untracked: data/Robocov_Box_all_genes.rda Untracked: data/Robocov_Precision_all_genes.rda Untracked: data/gene_names_GTEX_V6.txt Untracked: data/housekeeping_genes.txt Untracked: data/person_tissue_genes_voom.rda Untracked: docs/figure/ Untracked: sim_code/ Unstaged changes: Modified: .gitignore Modified: analysis/index.Rmd
library(Robocov)
library(corrplot)corrplot 0.84 loadedlibrary(ggplot2)Warning: package 'ggplot2' was built under R version 3.5.2Here we illustrare the Robocov estimates for a few genes of interest and then introduce the concept of Robospan.
robocov_gtex = get(load("/Users/kushaldey/Documents/Robocov-pages/data/Robocov_Box_all_genes.rda"))
dim(robocov_gtex)[1] 53 53 16069HBB (ENSG00000244734)
corrplot(robocov_gtex[,,"ENSG00000244734"], diag = TRUE,
col = colorRampPalette(c("blue", "white", "red"))(200),
tl.pos = "td", tl.cex = 0.4, tl.col = "black",
rect.col = "white",na.label.col = "white",
method = "color", type = "upper")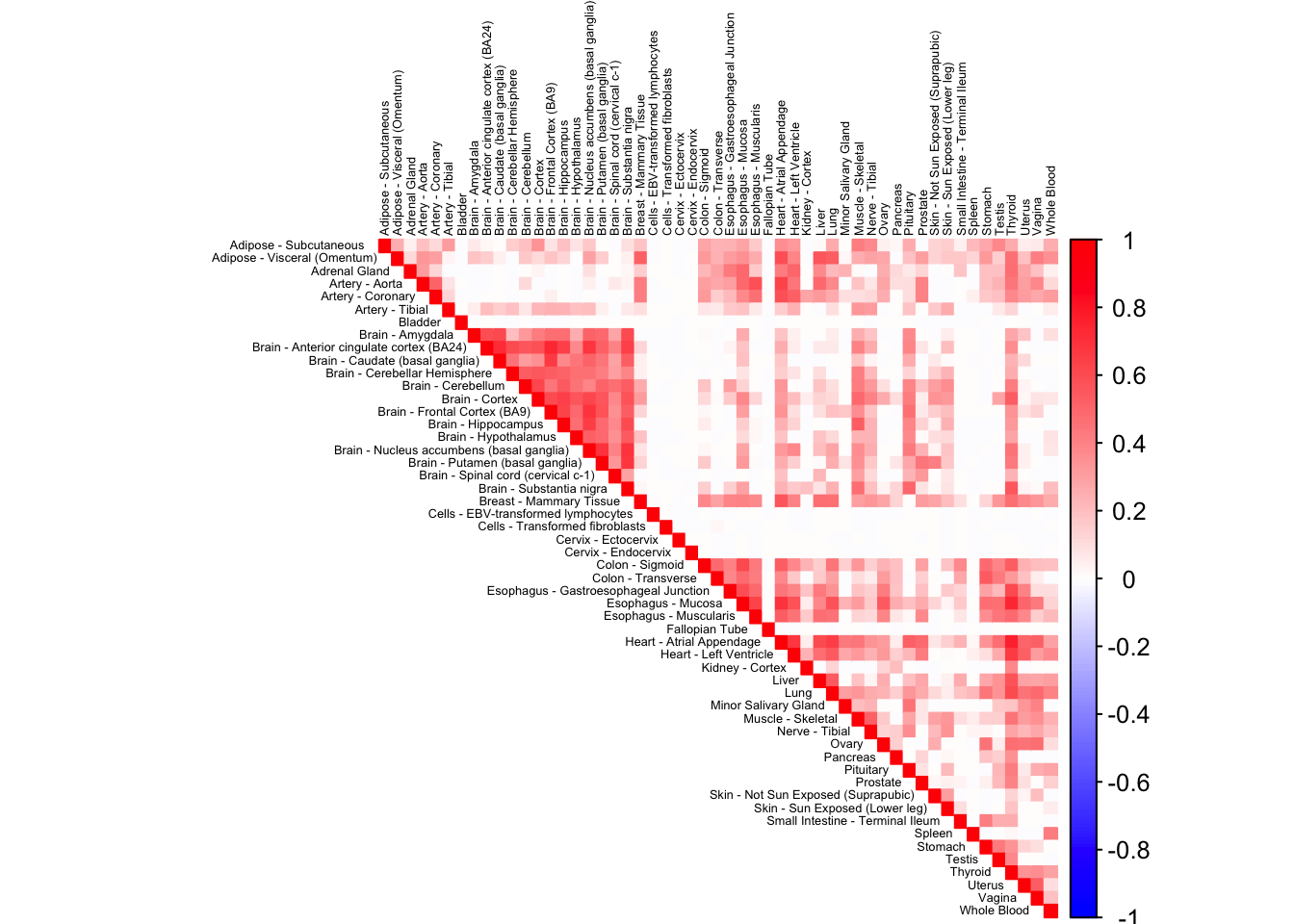
robospan = apply(robocov_gtex, 3, sum)/(53*53)
plot(density(robospan), xlab = "Robospan score", ylab = "density")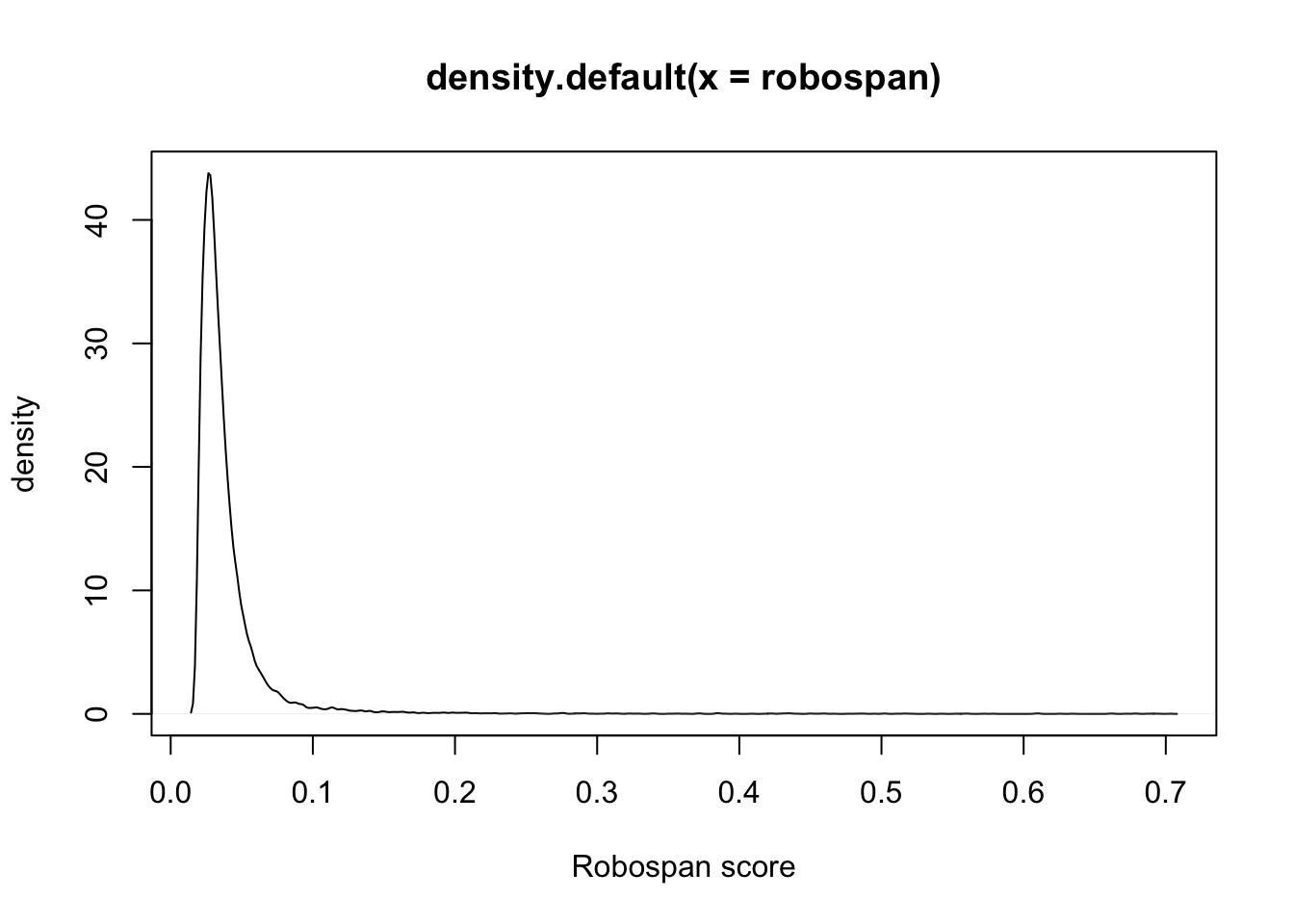
robospan[order(robospan, decreasing = T)[1:10]]ENSG00000134184 ENSG00000226278 ENSG00000184674 ENSG00000163682
0.7032426 0.6950521 0.6912778 0.6872412
ENSG00000233927 ENSG00000260246 ENSG00000204792 ENSG00000183298
0.6794906 0.6776832 0.6722359 0.6624388
ENSG00000196436 ENSG00000271581
0.6610709 0.6344899 robospan["ENSG00000244734"]ENSG00000244734
0.1639102 corrplot(robocov_gtex[,,"ENSG00000019991"], diag = TRUE,
col = colorRampPalette(c("blue", "white", "red"))(200),
tl.pos = "td", tl.cex = 0.4, tl.col = "black",
rect.col = "white",na.label.col = "white",
method = "color", type = "upper")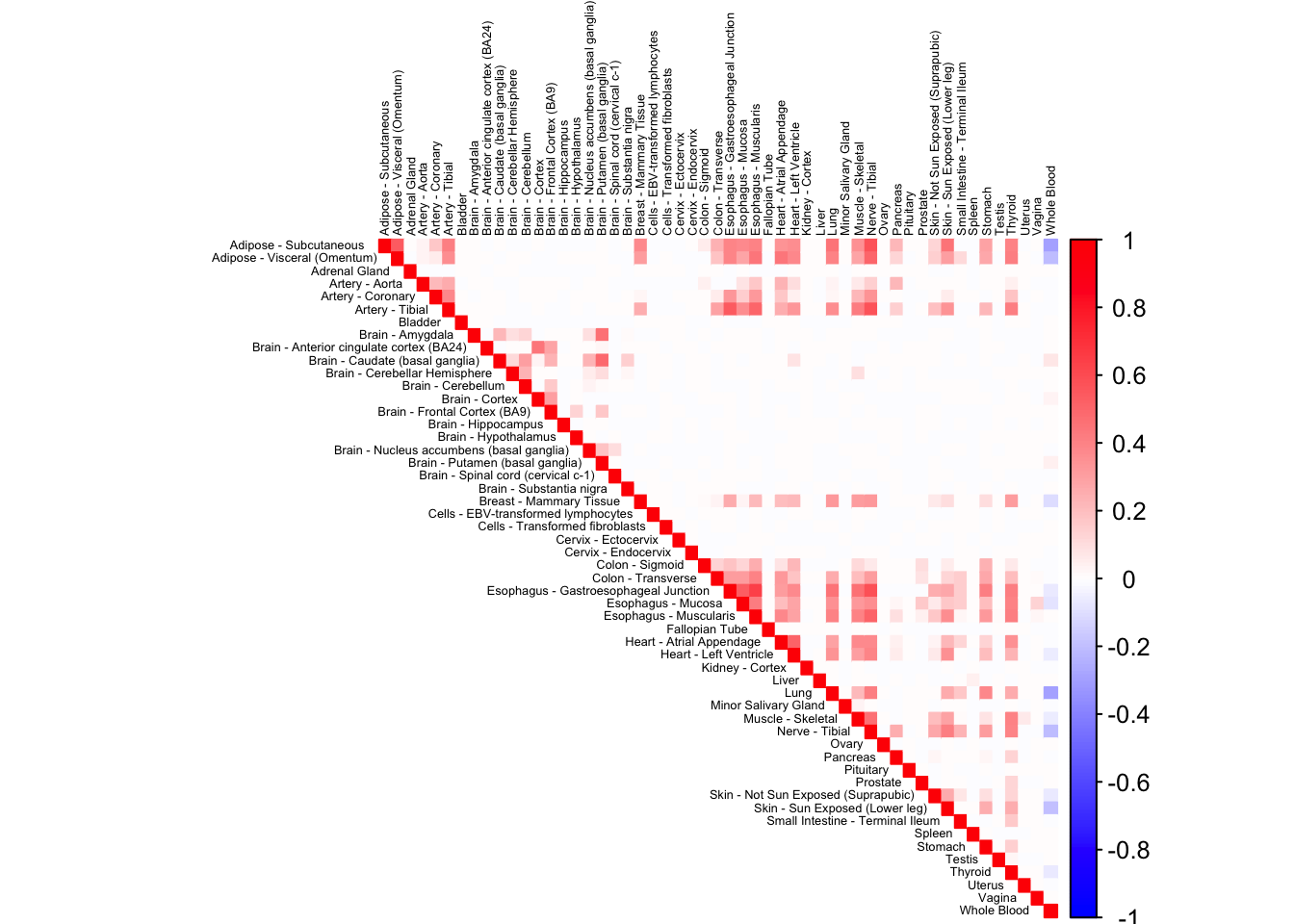 Ensembl and Gene symbol names
Ensembl and Gene symbol names
ensembl_gene_symbol = read.table("/Users/kushaldey/Documents/Robocov-pages/data/Gene_Scores/ensembl_and_hgnc_symbols.txt")
head(ensembl_gene_symbol) V1 V2
1 ENSG00000000419 DPM1
2 ENSG00000000457 SCYL3
3 ENSG00000000460 C1orf112
4 ENSG00000000938 FGR
5 ENSG00000000971 CFH
6 ENSG00000001036 FUCA2gene_symbols = ensembl_gene_symbol[match(names(robospan), ensembl_gene_symbol[,1]), 2]
robospan2 = robospan[which(!is.na(gene_symbols))]
names(robospan2) = gene_symbols[which(!is.na(gene_symbols))]Robospan mean
genes = names(robospan2)[order(robospan2, decreasing = T)[1:1600]]
df = cbind.data.frame(genes, 1)
write.table(df, file = "/Users/kushaldey/Documents/Robocov-pages/data/Gene_Scores/Robospan_mean.txt",
row.names = F, col.names = F, sep = "\t", quote=F)Robospan Num Up 0.1
robospan = apply(robocov_gtex, 3, function(x) length(which(x > 0.1)))/(53*53)
gene_symbols = ensembl_gene_symbol[match(names(robospan), ensembl_gene_symbol[,1]), 2]
robospan2 = robospan[which(!is.na(gene_symbols))]
names(robospan2) = gene_symbols[which(!is.na(gene_symbols))]
genes = names(robospan2)[order(robospan2, decreasing = T)[1:1600]]
df = cbind.data.frame(genes, 1)
write.table(df, file = "/Users/kushaldey/Documents/Robocov-pages/data/Gene_Scores/Robospan_Numbin_up_0_1.txt",
row.names = F, col.names = F, sep = "\t", quote=F)Robospan mean Blood
robospan = apply(robocov_gtex[,53,], 2, mean)
gene_symbols = ensembl_gene_symbol[match(names(robospan), ensembl_gene_symbol[,1]), 2]
robospan2 = robospan[which(!is.na(gene_symbols))]
names(robospan2) = gene_symbols[which(!is.na(gene_symbols))]
genes = names(robospan2)[order(robospan2, decreasing = T)[1:1600]]
df = cbind.data.frame(genes, 1)
write.table(df, file = "/Users/kushaldey/Documents/Robocov-pages/data/Gene_Scores/Robospan_Mean_Blood.txt",
row.names = F, col.names = F, sep = "\t", quote=F)Session information
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS High Sierra 10.13.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggplot2_3.1.1 corrplot_0.84 Robocov_0.1-6
loaded via a namespace (and not attached):
[1] gmp_0.5-13.2 Rcpp_1.0.1 pillar_1.3.1
[4] compiler_3.5.1 git2r_0.23.0 CVXR_0.99-2
[7] plyr_1.8.4 workflowr_1.1.1 R.methodsS3_1.7.1
[10] R.utils_2.7.0 tools_3.5.1 digest_0.6.19
[13] bit_1.1-14 tibble_2.1.1 evaluate_0.12
[16] gtable_0.3.0 lattice_0.20-35 pkgconfig_2.0.2
[19] rlang_0.4.2 Matrix_1.2-14 yaml_2.2.0
[22] withr_2.1.2 dplyr_0.8.0.1 Rmpfr_0.7-1
[25] ECOSolveR_0.4 stringr_1.4.0 knitr_1.20
[28] tidyselect_0.2.5 rprojroot_1.3-2 bit64_0.9-7
[31] grid_3.5.1 glue_1.3.1 R6_2.4.0
[34] rmarkdown_1.10 purrr_0.3.2 magrittr_1.5
[37] whisker_0.3-2 backports_1.1.4 scales_1.0.0
[40] htmltools_0.3.6 scs_1.1-1 assertthat_0.2.1
[43] colorspace_1.4-1 stringi_1.4.3 lazyeval_0.2.2
[46] munsell_0.5.0 crayon_1.3.4 R.oo_1.22.0 This reproducible R Markdown analysis was created with workflowr 1.1.1